G2.3 Distribusjon
Generelt
Distribusjon (fordeling) av legemidler karakteriseres både ved den hastigheten som legemidler transporteres fra blodbanen til vev med, og den fordelingen som finnes mellom ulike vev ved fordelingslikevekt. For enkelte legemidler skjer fordelingen så raskt at det tilsynelatende er fordelingslikevekt hele tiden. For de fleste legemidler sees en fordelingsfase etter absorpsjon før det inntrer fordelingslikevekt, se figur G2.1. I sistnevnte tilfelle vil det oppnådde maksimalnivå i plasmakonsentrasjonen etter dosering falle raskt i begynnelsen, fordi legemidlet da fordeles fra blodbanen og ut til vev (distribusjonsfase) samtidig som det elimineres. Etter at fordelingsfasen er avsluttet, vil legemiddelkonsentrasjonen falle langsommere (eliminasjonsfase). I denne fasen fordeles legemidlet tilbake til plasma etter hvert som lever og nyrer eliminerer legemidlet fra blodbanen.
Distribusjonsvolumet defineres enklest som forholdet mellom mengde legemiddel i kroppen og plasmakonsentrasjonen ved distribusjonslikevekt. Distribusjonsvolumet er en proporsjonalitetsfaktor som beskriver forholdet mellom mengde legemiddel i kroppen og plasmakonsentrasjon, se figur G2.2. Distribusjonsvolumet er en teoretisk størrelse som ikke tilsvarer noe fysiologisk rom i kroppen.
Lavest distribusjonsvolum har legemidler med høy binding til plasmaproteiner og lavt opptak/lav binding i vev (f.eks. heparin og warfarin). Verdier for distribusjonsvolumet som ikke er særlig mye større enn plasmavolumet kan da sees. Legemidler med høy binding i vev i forhold til binding til plasmaproteiner har store distribusjonsvolum (f.eks. citalopram og klorokin).
Distribusjon antas å skje hovedsakelig ved passiv diffusjon. Hastigheten av distribusjonen til et organ vil da være bestemt av legemiddelmolekylets molekylvekt, lipidløselighet, ioniseringsgrad, konsentrasjon i blodet og blodgjennomstrømningen i organet. I tillegg påvirkes distribusjonen av mange legemidler av aktiv transport over cellemembraner. Transportproteiner som tilhører ABC-klassen (ABC = ATP-binding cassette), som P-glykoprotein (Pgp), kan transportere legemidler ut av celler. Kreftceller kan på denne måten bli mer resistente mot cytostatika som har intracellulært angrepspunkt (metotreksat, vinkristin, antrasykliner). Annengenerasjons antihistaminer transporteres ut gjennom blod-hjerne-barrieren av Pgp. Dermed blir konsentrasjonen i sentralnervesystemet lav i forhold til i perifert vev, med mindre risiko for sedasjon. Også andre transportproteiner, både innenfor ABC-klassen og i klassene med organiske aniontransportører (OAT og OATP) og organiske kationtransportører (OCT) bidrar til å opprettholde en funksjonell blod-hjerne-barriere. Organiske aniontransportører er spesialiserte pumper som transporterer svake syrer, inklusive mange legemidler, ut av hjernen. Disse spesialiserte tranportproteinene finnes også mange andre steder i kroppen, inklusive i tarm, i lever/galleganger og i nyretubuli, der de pumper ut legemidler.
Binding av legemidler til plasmaproteiner påvirker kinetikk og effekt. Graden av proteinbinding angis ofte i prosent, men kan også angis som fri fraksjon, dvs. forholdet mellom fri konsentrasjon og totalkonsentrasjon i plasma. Dersom legemidlet ikke er bundet til plasmaproteiner, er fri fraksjon 1. Dersom legemidlet er sterkt bundet til plasmaproteiner (nær 100 % plasmaproteinbinding), vil fri fraksjon nærme seg 0. Det er den frie konsentrasjonen som reflekterer den farmakologiske aktiviteten av et legemiddel. Sure legemidler (fenytoin, valproat, salisylsyre m.fl.) bindes hovedsakelig til albumin, mens basiske legemidler (lidokain, propranolol, klozapin m.fl.) fortrinnsvis bindes til alfa-1-surt glykoprotein (AGP, orosomukoid). Nivåene av alfa-1-surt glykoprotein øker ved inflammasjon. Derfor kan mange basiske legemidler utvise stor variasjon i totalkonsentrasjonen i plasma avhengig av slike forhold, uten at konsentrasjonen av fritt legemiddel, og dermed heller ikke effekten, forandres nevneverdig. Serumkonsentrasjonsmålinger av legemidler kan være vanskelige å tolke ved tilstander som gir endringer i konsentrasjonen av eller bindingsaffiniteten for de plasmaproteiner som binder legemidlet. (Se også G3 Legemiddelbruk og -dosering ved nedsatt nyrefunksjon, G4 Legemiddeldosering ved mage- og tarmsykdom, leversykdom, hjertesykdom og nevrologisk sykdom og G16 Analyser av farmakologiske substanser og farmakogenetikk.)
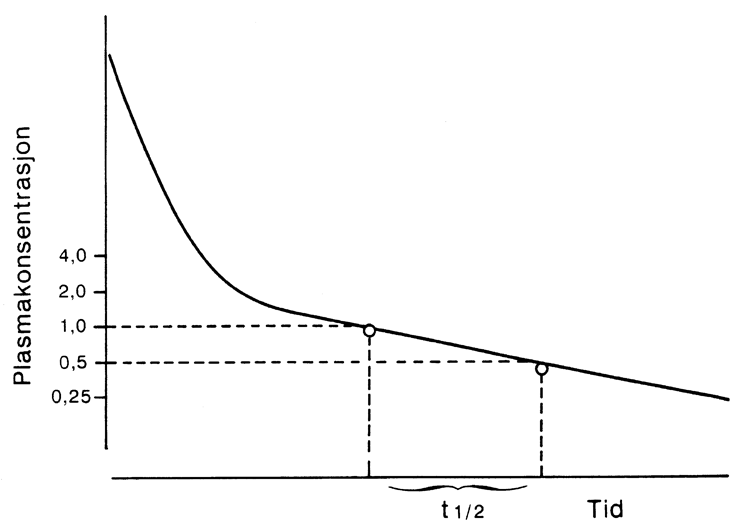
Figur G2.1 Plasmakonsentrasjons-tidskurve etter intravenøs injeksjon av et legemiddel.
Raskere fall i konsentrasjon i første del av kurven skyldes fordeling ut til vevene. Lineært fall (egentlig loglineært) i konsentrasjonen i eliminasjonsfasen (siste del av kurven) innebærer en konstant halveringstid (t1/2) og viser at eliminasjonen skjer ved førsteordenskinetikk. For legemidler som fordeler seg momentant til vevene, sees ikke noen egen fordelingsfase, og fallet i konsentrasjon er lineært fra starten. Merk at y-aksen er logaritmisk.
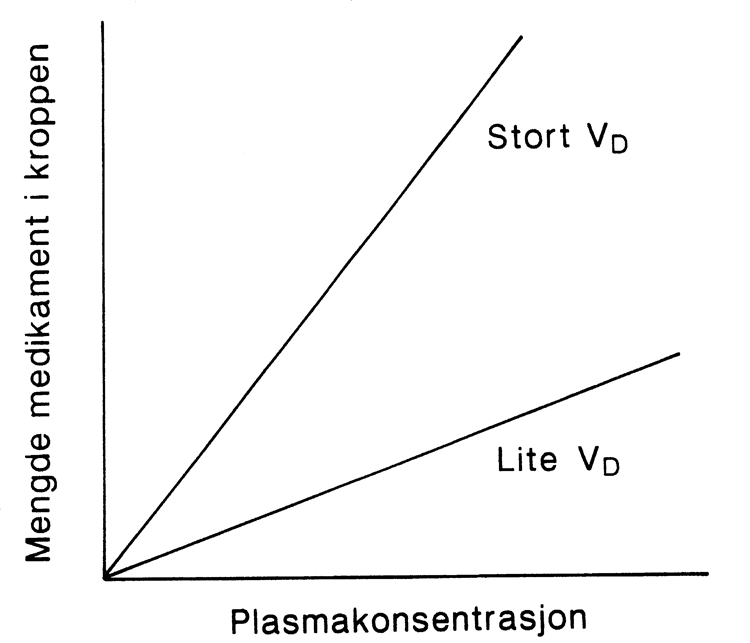
Figur G2.2 Mengde legemiddel i kroppen som funksjon av distribusjonsvolum og plasmakonsentrasjon.
Ved distribusjonslikevekt er det en lineær sammenheng mellom mengden legemiddel i kroppen (X) og plasmakonsentrasjonen (Cp) hvor distribusjonsvolumet (VD) representerer vinkelkoeffisienten: X = VD × Cp. Ved en gitt plasmakonsentrasjon vil den totale mengden i kroppen av et legemiddel med stort distribusjonsvolum være langt større enn av et legemiddel med lite distribusjonsvolum.